Pillar Wissen09.06.2026 · 11:00 Uhr9 Min Lesezeitvon Daniel Herrmann
CSA vs CSV: Was die FDA Final Guidance für Ihre Validierungsstrategie bedeutet
CSA vs CSV: direkter FDA-Scope, Prozessrisiko, geeignete Assurance-Aktivitäten und objektive Nachweise für Ihre Validierungsstrategie.
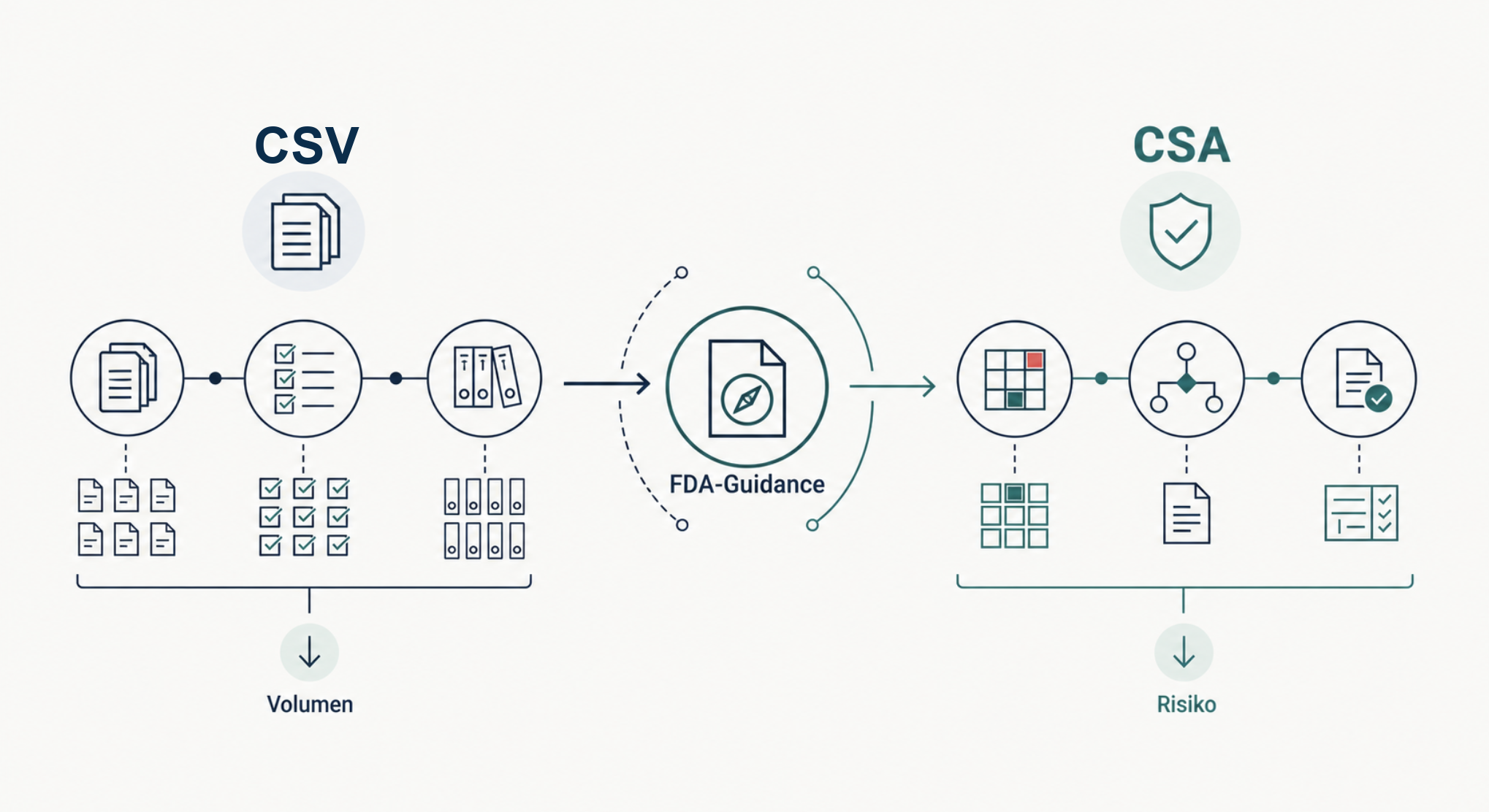
Computer Software Assurance (CSA) ist die finale FDA-Guidance für risikobasierte Software Assurance in der Medizinprodukteproduktion und in Quality Management Systems. Die im Februar 2026 revidierte Guidance ersetzt die Fassung vom September 2025 und richtet sie an der geänderten 21 CFR Part 820 Quality Management System Regulation (QMSR) aus. Für andere Pharma-GxP-Kontexte können die Methoden eine risikobasierte CSV unterstützen; der direkte FDA-Scope wird dadurch nicht erweitert.
Die FDA hat ihre finale Guidance „Computer Software Assurance for Production and Quality Management System Software” im Februar 2026 revidiert. Diese Fassung ersetzt die Final Guidance vom 24. September 2025 und berücksichtigt die Änderungen an 21 CFR Part 820 durch die Quality Management System Regulation (QMSR). Der inhaltliche Kern bleibt: Test-Volumen ist kein Synonym für Software Assurance. Für IT-Leiter, QA-Verantwortliche und Business Process Owner ist deshalb zuerst der Scope zu klären. Dieser Beitrag zeigt, was CSA von verbreiteter CSV-Praxis unterscheidet, wann die FDA-Guidance direkt greift und wie sich die risikobasierten Methoden sauber übertragen lassen.
Was CSA wirklich ist — die Definition der FDA
Computer Software Assurance ist die methodische Antwort der FDA auf eine seit Jahren wachsende Diskrepanz. Auf der einen Seite stand die etablierte CSV-Praxis: dokumentationsschwer, testintensiv, in der Tendenz formalistisch. Auf der anderen Seite stand die Realität moderner Software-Entwicklung: agil, iterativ, cloud-basiert, mit immer kürzeren Release-Zyklen. CSA schließt diese Lücke.
Der Scope ist klar umrissen: CSA gilt für Computer und automatisierte Datenverarbeitungssysteme, die in der Produktion von Medizinprodukten oder im zugehörigen Quality Management System eingesetzt werden. Dazu können je nach Intended Use etwa Produktions-, Labor-, Trainings-, Dokumenten- oder Qualitätsanwendungen gehören. Software as a Medical Device folgt eigenen regulatorischen Anforderungen. Ein LIMS, MES oder ERP-Modul fällt nicht allein wegen seines Produktnamens unter CSA; entscheidend sind Herstellerkontext und Intended Use.
Im Kern verschiebt CSA die Logik der Validierung: weg von „testen Sie alles, was technisch testbar ist”, hin zu „bewerten Sie risikobasiert, was die Patientensicherheit und Produktqualität schützt”. Diese Verschiebung ist keine Aufweichung der Anforderungen. Sie ist eine Präzisierung dessen, was Validierung leisten soll: Sicherheit für Patient und Produkt — nicht Vollständigkeit von Test-Skripten.
Wichtig für die Einordnung: CSA existiert parallel zu 21 CFR Part 11 und 21 CFR Part 820. Die regulatorischen Pflichten — Audit-Trails, elektronische Signaturen, Datenintegrität, Quality-System-Anforderungen — bleiben unverändert. CSA ändert nicht, was Sie nachweisen müssen. CSA ändert, wie Sie es nachweisen.
CSV vs CSA im direkten Vergleich
Die folgende Tabelle zeigt die zentralen Verschiebungen zwischen klassischer CSV-Praxis und der FDA-CSA-Methodik:
Aspekt
dokumentenlastige CSV-Praxis
CSA (FDA Final Guidance, revidiert 2026)
Grundansatz
Compliance-First
Assurance-First (Patient + Produkt)
Test-Philosophie
Volltestung, scripted
Risikobasiert, fokussiert
Dokumentations-Tiefe
Umfangreich, prozedural
Schlank, nutzenorientiert
Denkmodus
Beweise alles
risikobasierte Begründung
Validierungs-Effort
Hoch, oft proportional zur Funktionszahl
Proportional zum Risiko
Patient-Safety-Fokus
Implizit
Explizit, primär
Audit-Trail
Test-Volumen-orientiert
Beurteilungs-Begründung-orientiert
Modernisierungs-Anschluss
Schwer kompatibel mit Agile/DevOps
Nativ unterstützt
Die Vergleichstabelle zeigt: CSA ist nicht weniger als CSV — CSA ist anders strukturiert. Wer die zweite Spalte gegen die dritte liest und denkt „weniger Aufwand = weniger Sicherheit”, hat den Punkt verfehlt. CSA reduziert Redundanz, nicht Substanz. Die Audit-Belastbarkeit bleibt — sie entsteht nur durch dokumentierte Bewertung statt durch dokumentiertes Test-Volumen.
„CSA ist nicht weniger CSV. CSA ist klüger CSV.”
Die vier Kernprinzipien der FDA-Methodik
Für die Umsetzung verdichten wir die Guidance auf vier Arbeitsprinzipien. Das ist ein DHC-Praxis-Modell, keine wörtliche FDA-Gliederung. Es bildet einen handhabbaren Rahmen für Software Assurance im direkten CSA-Scope.
Erstes Prinzip: Intended Use definieren. Bevor validiert wird, muss klar sein, wofür die Software eingesetzt wird. Eine LIMS-Funktion, die Probenidentifikationen erzeugt, hat einen anderen Intended Use als eine LIMS-Funktion, die Sample-Labels druckt. Die FDA verlangt diese Differenzierung explizit — und nicht erst im Test-Skript, sondern bereits in der Validierungsplanung.
Zweites Prinzip: Risiko bewerten. Welche Funktionen können bei Fehlverhalten die Patientensicherheit oder Produktqualität beeinträchtigen? Diese Frage steuert den gesamten Validierungs-Aufwand. Hochrisiko-Funktionen werden ausführlich getestet. Niedrigrisiko-Funktionen werden — mit dokumentierter Begründung — reduziert oder über Lieferanten-Erklärungen abgedeckt. Die Risikobewertung ist kein Anhang; sie ist das Steuerungs-Instrument.
Drittes Prinzip: Assurance Activities planen. Risikobasiert wird entschieden, welche Aktivitäten den nötigen Vertrauensnachweis liefern: scripted oder unscripted Testing, Lieferanten-Nachweise, Monitoring und weitere geeignete Methoden. Die Auswahl muss zum Intended Use und Prozessrisiko passen; scripted Testing ist nicht pauschal vorgeschrieben.
Viertes Prinzip: Records angemessen führen. Die Dokumentation orientiert sich am Aufwand der Assurance Activities. Eine ausführlich getestete Hochrisiko-Funktion erhält vollständige Test-Records. Eine über Lieferanten-Bewertung abgedeckte Niedrigrisiko-Funktion erhält eine knappe Begründungs-Dokumentation. Die Idee: jede Seite Dokumentation hat einen Zweck. Was keinen Zweck hat, wird auch nicht produziert.
Diese vier Prinzipien wirken zusammen. Wer das erste vernachlässigt, kann das zweite nicht sauber bewerten. Wer das dritte falsch wählt, produziert beim vierten entweder zu wenig oder zu viel. CSA ist methodisch — und genau dadurch entsteht der Effizienzgewinn.
Wann CSA gilt — und wann weiterhin CSV
Die Frage „CSA oder CSV” ist nicht binär. Beide Methodiken bestehen 2026 parallel — mit klarem Anwendungsbereich:
CSA gilt direkt für: Computer und automatisierte Datenverarbeitungssysteme in der Medizinprodukteproduktion oder im Quality Management System eines Medizinprodukteherstellers. Ob LIMS, MES, ERP-, Dokumenten-, Trainings- oder Qualitätsanwendung in den Scope fällt, hängt vom konkreten Intended Use ab.
Andere CSV-Rahmen bleiben relevant für: pharmazeutische GMP-, GCP-, GLP- oder GDP-Systeme außerhalb dieses direkten Scopes sowie für andere regulatorische Kontexte. Risikobasierte Assurance kann dort nach GAMP 5, ICH Q9 und den jeweils anwendbaren Regeln umgesetzt werden, ohne die FDA-CSA-Guidance als unmittelbare Anforderung auszugeben.
Software als Medizinprodukt (SaMD) folgt einer eigenen regulatorischen Logik. 21 CFR Part 820, IEC 62304 und die jeweiligen FDA-Guidances zu AI/ML-SaMD bilden hier den Rahmen. CSA-Prinzipien sind methodisch verwandt, aber nicht direkt anwendbar.
Praktisch bedeutet das für Ihre IT-Landschaft: klassifizieren Sie zuerst Herstellerkontext, Intended Use und regulatorische Grundlage. Nur Systeme im direkten Scope werden als FDA-CSA-Anwendung geführt. Für andere GxP-Systeme können geeignete Methoden übernommen werden, müssen aber in ihrem eigenen Regelwerk begründet bleiben.
Was sich praktisch in Ihrer Validierungs-Strategie ändert
Wer die CSA-Methodik konsequent anwendet, sieht in drei Bereichen messbare Veränderungen.
Test-Volumen kann gezielt sinken. Wenn Prozessrisiko, Intended Use und verwertbare Lieferantennachweise es tragen, können pauschal geskriptete Tests durch passendere Assurance-Aktivitäten ersetzt werden. Das Ergebnis ist nicht automatisch weniger Arbeit, sondern ein besser begründeter Einsatz der Arbeit.
Dokumentations-Struktur verändert sich. CSV-Dokumentation ist klassischerweise vorlagengetrieben — jede Funktion wird nach demselben Schema beschrieben. CSA-Dokumentation ist begründungsgetrieben — jede Funktion wird nach ihrem Risiko-Profil dokumentiert. Vorlagen bleiben hilfreich, aber sie werden zu Werkzeugen, nicht zu Pflicht-Strukturen.
Die Begründung wird zum zentralen Nachweis. Ihr Dossier muss zeigen, warum eine Funktion als hoch oder nicht hoch im Prozessrisiko bewertet wurde und weshalb die gewählten Assurance-Aktivitäten angemessen sind. Audit-Vorbereitung fokussiert deshalb nicht nur Testnachweise, sondern auch den nachvollziehbaren Entscheidungspfad.
Konkret heißt das für Ihre Validierungsstrategie: Anwendbarkeit klären, Risikobewertungen und Teststrategien neu ausrichten, Reviewer-Kompetenz aufbauen und betroffene Verfahren anpassen. Zeitplan und Aufwand hängen von Portfolio, Governance, Systemrisiko und verfügbarem Nachweisbestand ab.
Der Weg zur Implementierung — wo Sie anfangen
CSA-Einführung ist kein Big-Bang-Projekt. Sie ist ein strukturierter Übergang in vier Schritten:
System-Bestand klassifizieren. Welche Ihrer GxP-Systeme fallen unter CSA-Anwendungsbereich? Welche nicht? Eine einfache Klassifikations-Tabelle reicht als Startpunkt.
CSA-Pilotsystem auswählen. Beginnen Sie mit einem klar abgegrenzten System im direkten CSA-Scope, dessen Prozessrisiko und Nachweisbestand eine belastbare Pilotbewertung erlauben.
Methodik im Pilot anwenden. Risikobewertung, Assurance Activities, angemessene Records — die vier Prinzipien im Pilot durchziehen. Dokumentieren Sie, was funktioniert und was anzupassen ist.
Framework skalieren. Auf Basis des Piloten passen Sie Ihr SOP-Set, Ihre Vorlagen und Ihre Reviewer-Schulung an. Dann rollen Sie die Methodik auf das gesamte System-Portfolio aus.
Der Zeitplan wird nach Anzahl und Kritikalität der Systeme, betroffenen Verfahren, Reviewer-Kompetenz und Governance-Reife festgelegt. Der Pilot liefert die belastbare Grundlage für den Rollout. Für die strukturierte Übertragung auf Ihr Portfolio bietet unsere CSV-Beratung den passenden Rahmen.
Häufige Fragen
Ersetzt CSA das CSV-Framework vollständig?
Nein. CSA beschreibt im direkten Scope einen Ansatz für Software Assurance; die Pflicht zum Eignungsnachweis bleibt bestehen. Außerhalb der Medizinprodukteproduktion und des zugehörigen Quality Management Systems gelten die jeweils anwendbaren CSV- und GxP-Anforderungen. 21 CFR Part 11 und die Anforderungen des Quality Management Systems bleiben unberührt.
Gilt CSA auch außerhalb der USA, etwa für EU-Pharma?
CSA ist eine FDA-Guidance mit einem definierten US-regulatorischen Scope. Risikobasierte Methoden sind auch mit europäischen GxP-Ansätzen anschlussfähig, daraus entsteht jedoch keine unmittelbare CSA-Pflicht für EU-Pharma. Die anwendbare Rechts- und Guidance-Basis muss pro System festgelegt werden.
Wie verhalten sich CSA und GAMP 5 2nd Edition zueinander?
Sie können sich ergänzen, sind aber nicht austauschbar. GAMP 5 Second Edition ist breiter und unterstützt risikobasierte Validierung in unterschiedlichen GxP-Kontexten. FDA CSA gilt direkt für Software in der Medizinprodukteproduktion und in Quality Management Systems. Methodische Übereinstimmungen helfen bei der Umsetzung; die Erfüllung beider Referenzen muss dennoch separat begründet werden.
Daniel Herrmann Consulting — Boutique-Beratung für GxP-Compliance und Computer System Validation in Pharma, Biotech und MedTech. 15+ Jahre Hands-on-Expertise. 60+ validierte Systeme. 100 % Audit-Bestehensquote. 0 kritische Findings.
Gemäß § 36 VSBG: Wir sind nicht bereit oder verpflichtet, an Streitbeilegungsverfahren vor einer Verbraucherschlichtungsstelle teilzunehmen.
Haftung für Inhalte
Wir sind für eigene Inhalte auf diesen Seiten nach den allgemeinen Gesetzen verantwortlich. Nach den Artikeln 4 bis 8 der Verordnung (EU) 2022/2065 (Digital Services Act, DSA) sind wir als Diensteanbieter jedoch nicht verpflichtet, übermittelte oder gespeicherte fremde Informationen zu überwachen oder nach Umständen zu forschen, die auf eine rechtswidrige Tätigkeit hinweisen.
Hinweis: Die §§ 7–10 des vormaligen Telemediengesetzes (TMG) zur Haftung digitaler Dienste wurden — entgegen einer verbreiteten Übergangsformel — nicht in das Digitale-Dienste-Gesetz (DDG) übernommen. Die Haftung für digitale Dienste richtet sich seit dem 14. Mai 2024 maßgeblich nach der Verordnung (EU) 2022/2065 (DSA), insbesondere nach den Artikeln 4–8 — die wir oben referenzieren. Die §§ 7–10 DDG haben andere Regelungsinhalte (beschränkte Verantwortlichkeit mit Verweis auf DSA Art. 4–8 und WLAN-Zugänge, Sperrungsansprüche bei Urheberrechts-Verletzungen, Listen der Anbieter audiovisueller Mediendienste und Videosharing-Plattformen sowie Auskunftsverlangen der nach Landesrecht zuständigen Behörden).
Urheberrecht
Die durch die Seitenbetreiber erstellten Inhalte und Werke auf diesen Seiten unterliegen dem deutschen Urheberrecht. Die Vervielfältigung, Bearbeitung, Verbreitung und jede Art der Verwertung außerhalb der Grenzen des Urheberrechtes bedürfen der schriftlichen Zustimmung des jeweiligen Autors.
Datenschutzerklärung
1. Datenschutz auf einen Blick
Die folgenden Hinweise geben einen einfachen Überblick darüber, was mit Ihren personenbezogenen Daten passiert, wenn Sie diese Website besuchen. Personenbezogene Daten sind alle Daten, mit denen Sie persönlich identifiziert werden können.
2. Verantwortlicher
Verantwortlicher im Sinne des Art. 4 Nr. 7 DSGVO für die Datenverarbeitung auf dieser Website ist:
Daniel Herrmann Consulting
Daniel Herrmann (Einzelunternehmer)
Enzweilerweg 3a · 66709 Weiskirchen · Deutschland
Telefon: +49 170 7878065
E-Mail: info@daniel-herrmann.io
Ein Datenschutzbeauftragter ist nicht bestellt; Ansprechpartner in allen Datenschutzfragen ist der Verantwortliche.
3. Server-Logs (technisch notwendig)
Beim Besuch dieser Website werden vom Hosting-Anbieter automatisch technische Informationen in Server-Logfiles erfasst: IP-Adresse (nach 7 Tagen anonymisiert), Datum und Uhrzeit, aufgerufene Seite, Referrer, User Agent. Rechtsgrundlage: Art. 6 Abs. 1 lit. f DSGVO — berechtigtes Interesse am technisch fehlerfreien Betrieb. Speicherdauer: 7 Tage, anschließend automatische Löschung. Eine Zusammenführung mit anderen Datenquellen findet nicht statt.
4. Kontaktaufnahme
Wenn Sie uns per Formular, E-Mail oder Telefon kontaktieren, speichern wir Ihre Angaben (Name, E-Mail, Unternehmen, Nachrichteninhalt) zur Bearbeitung der Anfrage und für Anschlussfragen. Rechtsgrundlage: Art. 6 Abs. 1 lit. b DSGVO (vorvertragliche Maßnahmen) und Art. 6 Abs. 1 lit. f DSGVO (berechtigtes Interesse an der Beantwortung von Anfragen). Speicherdauer: Löschung, sobald die Anfrage abschließend bearbeitet ist, spätestens nach 12 Monaten, sofern keine gesetzlichen Aufbewahrungspflichten entgegenstehen. Eine Weitergabe an Dritte erfolgt nicht.
5. Lead-Magnets (PDF-Downloads)
Zur Anforderung unserer Praxis-Guides (z. B. Strategie-Guide, Compliance-Gap-Analyse, Execution-Playbook) bitten wir um Ihre E-Mail-Adresse sowie optional um Name und Unternehmen. Die Angaben werden ausschließlich zur Bereitstellung des angeforderten Materials und zur Dokumentation der Einwilligung gespeichert. Rechtsgrundlage: Art. 6 Abs. 1 lit. a DSGVO (Einwilligung). Speicherdauer: bis zu Ihrem Widerruf, maximal 24 Monate. Die Newsletter-Anmeldung wird als separate, opt-in-Checkbox auf dem Lead-Magnet-Formular angeboten (keine Kopplung — Sie können das Material auch ohne Newsletter-Abo anfordern). Ohne gesonderte Einwilligung erfolgt kein Marketing-Versand.
5a. Newsletter (Double-Opt-In)
Verfahren: Wenn Sie unseren Newsletter abonnieren, verwenden wir das Double-Opt-In-Verfahren: Nach Eingabe Ihrer E-Mail-Adresse und Ihres Namens erhalten Sie eine Bestätigungs-Mail mit Verifizierungs-Link. Erst nach Klick auf diesen Link aktivieren wir das Abonnement und dokumentieren Ihre Einwilligung. Daten: E-Mail-Adresse, Name, Zeitstempel der Einwilligung, Confirmation-IP. Zweck: Versand des monatlichen dhc-Newsletters mit Praxis-Insights und Dokumentation des Einwilligungs-Vorgangs gem. Art. 7 Abs. 1 DSGVO. Rechtsgrundlage: Art. 6 Abs. 1 lit. a DSGVO (Einwilligung), § 7 Abs. 2 Nr. 3 UWG. Speicherdauer: für die Dauer Ihres aktiven Abonnements; Einwilligungs-Logs werden zusätzlich 3 Jahre nach Beendigung des Abonnements als Nachweis im Beschwerdefall vorgehalten. Widerruf: Jeder Newsletter enthält im Footer einen Ein-Klick-Abmelde-Link. Alternativ genügt eine kurze E-Mail an info@daniel-herrmann.io. Auftragsverarbeiter: Der Newsletter wird derzeit über die SMTP-Infrastruktur unseres Hosting-Anbieters versendet; wir setzen aktuell kein externes E-Mail-Marketing-Tool ein. Sobald wir auf ein dediziertes Newsletter-Tool (z. B. Brevo) wechseln, aktualisieren wir diesen Abschnitt und schließen einen ergänzenden Auftragsverarbeitungsvertrag.
5c. Link-Click-Tracking in unseren E-Mails
Verfahren: Um zu verstehen, welche Inhalte in unseren Lead-Magnet-Mails und Newsletter-Ausgaben tatsächlich nützlich sind, leiten wir ausgehende Links über einen internen Redirect-Endpunkt (/t/<token>). Jeder Link in jedem Versand erhält einen aggregierten Click-Zähler — wir speichern jedoch KEINE Empfänger-Kennung, KEINE IP-Adresse und KEINEN User-Agent. Die gespeicherten Daten entsprechen anonymen Server-Logs (z. B.: „Link X im Versand Y wurde insgesamt 42 mal geklickt") und lassen keinen Rückschluss zu, wer geklickt hat. Rechtsgrundlage: Da kein Personenbezug besteht (Erwägungsgrund 26 DSGVO — anonyme Daten, keine Verarbeitung personenbezogener Daten i. S. v. Art. 4 Abs. 1 DSGVO), ist keine gesonderte Einwilligung erforderlich. Für den Redirect-Vorgang selbst stützen wir uns auf Art. 6 Abs. 1 lit. f DSGVO (berechtigtes Interesse an der Messung der Inhalts-Relevanz unserer eigenen Kommunikation). Ausnahmen: Rechtlich relevante Links (Impressum, Datenschutz, Abmelde-Link) werden nicht über den Redirect geleitet — der Click auf solche Links wird nie gezählt. Speicherdauer: Die anonymen Counter werden 24 Monate aufbewahrt und danach gelöscht.
5b. Speicherdauer-Übersicht & Begründungen
Konsolidierte Übersicht aller Speicherdauern mit Begründung — das Prinzip: minimale Speicherung, klarer Zweck, dokumentierte Grundlage.
DatenkategorieSpeicherdauerBegründung
Server-Logs7 TageAusreichend für technische Fehleranalyse & Sicherheits-Forensik — Art. 6 Abs. 1 lit. f DSGVO. IP-Anonymisierung greift sofort beim Log-Schluss.
Kontaktanfragen12 MonateB2B-Sales-Zyklen in Pharma laufen typisch 6–9 Monate. 12 Monate deckt Anschluss-Anfragen ohne unnötige Vorhaltung. Gesetzliche Aufbewahrungspflichten (z. B. § 257 HGB) greifen erst bei Vertragsabschluss.
Lead-Magnetsbis zu 24 MonateBis Widerruf der Einwilligung; 24-Monats-Kappung deckt die Dokumentation der Einwilligung (Art. 7 DSGVO) und einen typischen Re-Engagement-Zyklus ab. Widerruf jederzeit, Löschung binnen 7 Tagen ab Eingang.
Newsletter-AboAbo-Dauer + 3 J. Consent-LogE-Mail + Name nur solange das Abo aktiv ist. Consent-Log (Zeitstempel + IP) wird 3 Jahre vorgehalten, um die Verjährungsfrist (§ 195 BGB) für Beschwerden nach § 7 UWG abzudecken.
Cookies (inkl. Analyse)30 Min – 14 MonatePro Tool detailliert in 6.9. GA4 ist auf 14 Monate gekappt (GA4-Minimum, kürzer nur über Property-Reset möglich); alle anderen Tools liegen unterhalb oder auf Branchen-Standard.
Cookiebot-Einwilligung12 MonateMaximum-Fenster, das die EDPB für wiederholte Einwilligungs-Abfragen für angemessen hält. Nach 12 Monaten erscheint das Banner erneut.
6. Cookies, Analyse & Tracking
Auf dieser Website kommen Analyse- und Tracking-Werkzeuge zum Einsatz, die über eine reine Reichweitenmessung hinausgehen und Nutzungsprofile erstellen. Dabei werden personenbezogene Daten (insb. IP-Adresse, Geräte- und Browser-Informationen, Verhaltensdaten) verarbeitet und in Drittländer (u. a. USA) übermittelt. Rechtsgrundlage ist Ihre Einwilligung gemäß § 25 Abs. 1 TTDSG und Art. 6 Abs. 1 lit. a DSGVO. Die Einwilligung erteilen Sie beim ersten Besuch über unser Cookie-Banner und können sie jederzeit mit Wirkung für die Zukunft widerrufen — siehe Abschnitt 6.10. Der Widerruf ist so einfach wie die Erteilung der Einwilligung (Art. 7 Abs. 3 DSGVO); die Rechtmäßigkeit der vor dem Widerruf erfolgten Verarbeitung bleibt unberührt.
6.0 Einwilligungsverwaltung (Cookiebot)
Anbieter: Cybot A/S, Havnegade 39, 1058 Kopenhagen, Dänemark (Tochter der Usercentrics-Gruppe) — EU-Anbieter. Zweck: Cookiebot ist die Consent-Management-Plattform (CMP), mit der wir Ihre Einwilligung im Sinne des § 25 TTDSG und Art. 7 DSGVO dokumentieren und Analyse- und Tracking-Tools bis zu Ihrer Zustimmung blockieren. Daten: Cookiebot setzt das Cookie „CookieConsent“ mit Ihrem Zustimmungsstatus (Kategorien, Zeitstempel, anonyme Kennung) sowie ein serverseitiges Consent-Log. Speicherdauer: bis zu 12 Monate ab Erteilung der Einwilligung; danach erneute Abfrage beim nächsten Besuch. Rechtsgrundlage: Die Verarbeitung der Einwilligungsdaten selbst stützt sich auf Art. 6 Abs. 1 lit. c DSGVO (gesetzliche Pflicht zur Dokumentation der Einwilligung). Drittlandübermittlung: primäre Verarbeitung in der EU; Unterauftragsverarbeiter können Dienstleister außerhalb des EWR unter EU-Standardvertragsklauseln umfassen. Auftragsverarbeitung: AV-Vertrag mit Cybot A/S geschlossen. Mehr Informationen:Datenschutzerklärung Cookiebot.
6.1 Google Analytics 4
Anbieter: Google Ireland Limited, Gordon House, Barrow Street, Dublin 4, Irland (Mutterkonzern: Google LLC, USA). Zweck: Tracking-Tool, das über eine reine Reichweitenmessung hinausgeht. Google nutzt die erhobenen Daten für den Betrieb von GA4 sowie teilweise für eigene Zwecke (insoweit keine reine Auftragsverarbeitung). Daten: pseudonyme Kennungen (Client-ID), IP-Adresse (auf EU-Server gekürzt, siehe unten), Seitenaufrufe, Verweildauer, Scroll-Tiefe, Klick-Events, Geräte- und Browser-Informationen, ungefährer Standort (Land / Region aus IP). Speicherdauer: bis zu 14 Monate auf Event-Ebene, danach automatische Löschung. IP-Kürzung: Die IP-Adresse wird auf einem EU-Server gekürzt, bevor sie in die USA weitergeleitet wird (das „_anonymizeIp“-Pendant in GA4 ist standardmäßig aktiv). Drittlandübermittlung: Verarbeitung auf Servern in den USA. Garantien: EU-Standardvertragsklauseln und das EU-US Data Privacy Framework (Angemessenheitsbeschluss vom 10. Juli 2023). Hinweis: Aufsichtsbehörden weisen darauf hin, dass die gewonnene Rechtssicherheit nur vorübergehender Natur sein könnte; die Vorgängerregelungen (Safe Harbor, Privacy Shield) wurden vom EuGH für unwirksam erklärt. Auftragsverarbeitung: AV-Vertrag mit Google Ireland Ltd. geschlossen. Mehr Informationen:Datenschutzerklärung Google, Browser-Opt-out.
6.2 Microsoft Clarity
Anbieter: Microsoft Ireland Operations Ltd., One Microsoft Place, South County Business Park, Leopardstown, Dublin 18, Irland (Mutterkonzern: Microsoft Corporation, USA). Zweck: Sitzungsaufzeichnungen und Heatmaps zur Verbesserung der Nutzerfreundlichkeit. Hinweis: Sitzungsaufzeichnungen sind eine besonders intensive Verarbeitungsform — sie können auch bei standardmäßiger Maskierung von Eingabefeldern sensible Inhalte erfassen. Wir haben die strengste Maskierungsstufe („Strict“) für alle Formularfelder konfiguriert. Daten: Mausbewegungen, Klicks, Scroll-Verhalten, Seitenaufrufe, Geräte- und Browser-Informationen, IP-Adresse (gekürzt), Land. Speicherdauer: bis zu 12 Monate ab der letzten erfassten Sitzung. Drittlandübermittlung: Microsoft ist ein US-Konzern, Daten können auf Servern in den USA verarbeitet werden. Garantien: EU-Standardvertragsklauseln und EU-US Data Privacy Framework. Die oben unter GA4 genannten Vorbehalte gelten sinngemäß. Auftragsverarbeitung: Microsoft Online Services DPA geschlossen. Mehr Informationen:Microsoft Datenschutzerklärung.
6.3 Leadfeeder (Dealfront)
Anbieter: Dealfront Germany GmbH (Leadfeeder), Markgrafenstraße 36, 10117 Berlin — europäischer Anbieter. Zweck: B2B-Identifikation von besuchenden Unternehmen anhand kommerziell lizenzierter IP-Adress-Datenbanken (u. a. RIPE, ARIN, öffentliche Unternehmens-IP-Bereiche) zur Information unserer Vertriebsansprache. Ziel ist die Identifikation des Unternehmens, nicht des einzelnen Nutzers. Wichtiger datenschutzrechtlicher Hinweis: IP-Adressen können personenbezogene Daten darstellen, insbesondere wenn sie mit anderen Daten verknüpft werden. Wir behandeln die Verarbeitung daher als DSGVO-relevant. Daten: IP-Adresse, Seitenaufrufe, Zeitstempel, Verweildauer, Referrer. Datenquellen: Dealfront reichert IP-Daten mit öffentlich zugänglichen Unternehmensinformationen und lizenzierten B2B-Datenbanken an. Speicherdauer: bis zu 12 Monate auf Besuchs-Ebene; aggregierte Reports können länger aufbewahrt werden. Drittlandübermittlung: Dealfront verarbeitet primär auf EU-Infrastruktur; Unterauftragsverarbeiter können US-Dienstleister umfassen, abgesichert über EU-Standardvertragsklauseln. Auftragsverarbeitung: AV-Vertrag mit Dealfront Germany GmbH geschlossen. Mehr Informationen:Datenschutzerklärung Dealfront.
6.4 Google Ads (Conversion-Tracking & Remarketing)
Anbieter: Google Ireland Limited, Gordon House, Barrow Street, Dublin 4, Irland (Mutterkonzern: Google LLC, USA). Zweck: Wir setzen Google Ads Conversion-Tracking sowie (sofern aktiviert) Remarketing ein, um den Erfolg von Werbekampagnen zu messen und Besucher mit relevanten Anzeigen anzusprechen. Daten: pseudonyme Click-ID (gclid), Conversion-Event, Conversion-Zeitstempel, Geräte- und Browser-Informationen. Eingesetzte Cookies u. a. „_gcl_au“ (Conversion-Linker) und „test_cookie“ (technisches Prüf-Cookie von doubleclick.net). Speicherdauer: „_gcl_au“ bis zu 90 Tage; Conversion-Logs im Google-Ads-Konto gemäß Google-Aufbewahrungseinstellungen. Drittlandübermittlung: Verarbeitung auf Google-Servern auch in den USA. Garantien: EU-Standardvertragsklauseln und EU-US Data Privacy Framework. Auftragsverarbeitung: AV-Vertrag mit Google Ireland Ltd. geschlossen. Mehr Informationen:Datenschutzerklärung Google, Werbe-Einstellungen anpassen.
6.5 LinkedIn Insight Tag
Anbieter: LinkedIn Ireland Unlimited Company, Wilton Plaza, Wilton Place, Dublin 2, Irland (Mutterkonzern: LinkedIn Corporation, USA). Zweck: Der LinkedIn Insight Tag ermöglicht Kampagnen-Messung, Zielgruppen-Analyse und (sofern aktiviert) Retargeting für LinkedIn-Werbekampagnen. Daten: IP-Adresse (gekürzt), Zeitstempel, Seiten-URL, Geräte-Merkmale, LinkedIn-Mitglieds-ID, sofern der Besucher bei LinkedIn eingeloggt war. Eingesetzte Cookies u. a. „bcookie“, „lidc“, „bscookie“. Speicherdauer: bis zu 6 Monate für direkt-personenbezogene Kennungen; aggregierte Kampagnen-Reports können länger aufbewahrt werden. Drittlandübermittlung: Verarbeitung auf LinkedIn-Servern auch in den USA. Garantien: EU-Standardvertragsklauseln und EU-US Data Privacy Framework. Auftragsverarbeitung: AV-Vertrag mit LinkedIn Ireland Unlimited Company geschlossen. Mehr Informationen:LinkedIn Datenschutzerklärung, LinkedIn-Opt-out.
6.5a Meta-Pixel (Facebook-Pixel)
Anbieter: Meta Platforms Ireland Ltd., Merrion Road, Dublin 4, D04 X2K5, Irland (Mutterkonzern: Meta Platforms, Inc., 1601 Willow Road, Menlo Park, CA 94025, USA). Zweck: Der Meta-Pixel (auch „Facebook-Pixel“) ermöglicht Reichweitenmessung, Conversion-Tracking und (sofern aktiviert) Retargeting für Werbekampagnen auf Facebook und Instagram. So können wir die Wirksamkeit unserer Anzeigen messen und Besuchern relevante Werbung ausspielen. Daten: pseudonyme Kennungen, IP-Adresse, Geräte- und Browser-Informationen, Referrer-URL, ausgelöste Events (z. B. Seitenaufruf). Eingesetzte Cookies u. a. „_fbp“ (First-Party-Cookie zur Wiedererkennung des Browsers) und „_fbc“ (Klick-Kennung aus dem URL-Parameter „fbclid“). War der Besucher zum Zeitpunkt des Besuchs bei Facebook / Instagram eingeloggt, kann Meta den Besuch dem jeweiligen Konto zuordnen. Gemeinsame Verantwortlichkeit: Für die Erhebung und Übermittlung der Daten an Meta besteht eine gemeinsame Verantwortlichkeit im Sinne des Art. 26 DSGVO zwischen uns und Meta; wir haben hierzu die von Meta bereitgestellte Vereinbarung zur gemeinsamen Verantwortlichkeit geschlossen. Für die anschließende Weiterverarbeitung der Daten durch Meta zu eigenen Zwecken ist Meta allein verantwortlich. Speicherdauer: „_fbp“ bis zu 90 Tage; auf Event-Ebene gemäß den Aufbewahrungseinstellungen im Meta Events Manager. Drittlandübermittlung: Verarbeitung auf Meta-Servern auch in den USA. Garantien: EU-Standardvertragsklauseln und EU-US Data Privacy Framework (Angemessenheitsbeschluss vom 10. Juli 2023). Die oben unter GA4 genannten Vorbehalte gelten sinngemäß. Rechtsgrundlage: Ihre Einwilligung gemäß § 25 Abs. 1 TTDSG und Art. 6 Abs. 1 lit. a DSGVO. Der Pixel wird erst nach Ihrer Einwilligung über unser Cookiebot-Banner geladen. Mehr Informationen:Datenschutzrichtlinie von Meta, Meta Werbe-Einstellungen.
6.6 Google Tag Manager
Anbieter: Google Ireland Limited (siehe 6.1). Zweck: Google Tag Manager (GTM) ist ein Tag-Management-System, das auf dieser Website andere Tracking-Tags (z. B. Google Analytics, Google Ads, LinkedIn Insight Tag) lädt. GTM selbst setzt keine Cookies und erhebt keine personenbezogenen Daten im engeren Sinne; es orchestriert lediglich die nach Einwilligung geladenen Tags. Daten: Technische Request-Daten (IP-Adresse, Zeitstempel, User Agent) werden kurzzeitig vom GTM-Loader-Server bei Google verarbeitet. GTM selbst setzt keinen persistenten Identifier. Drittlandübermittlung: Der GTM-Loader wird auf Google-Infrastruktur betrieben, auch in den USA. Garantien wie unter 6.1. Hinweis: Wir haben GTM so konfiguriert, dass kein Messtag ausgelöst wird, bevor Sie über unser Cookiebot-Banner eingewilligt haben. Mehr Informationen:Datenschutzerklärung Google.
6.7 Cloudflare (CDN & Bot-Management)
Anbieter: Cloudflare, Inc., 101 Townsend Street, San Francisco, CA 94107, USA, mit EU-Vertreter Cloudflare Germany GmbH, Rosental 7, c/o Mindspace, 80331 München. Zweck: Cloudflare wird von unserem Hosting-Anbieter als Content Delivery Network (CDN) und Security-Layer eingesetzt. Das Cookie „__cf_bm“ wird von Cloudflares Bot-Management gesetzt, um in Echtzeit Menschen von automatisiertem Traffic zu unterscheiden und DDoS-Angriffe sowie Content-Scraping abzuwehren. Warum wir das als technisch erforderlich einordnen: Ohne Bot-Mitigation wäre unsere Website angreifbar für Credential-Stuffing, Scraping und DDoS-Angriffe, die Verfügbarkeit, Integrität und die Sicherheit der von uns verarbeiteten personenbezogenen Daten (z. B. Lead-Magnet-Formulare, Newsletter-Anmeldungen) gefährden würden. Bot-Mitigation ist daher eine technische Sicherheitsmaßnahme im Sinne von Art. 32 DSGVO (Sicherheit der Verarbeitung). Cloudflare selbst klassifiziert „__cf_bm“ als „strictly necessary“ (<a href="https://developers.cloudflare.com/fundamentals/reference/policies-compliances/cloudflare-cookies/" target="_blank" rel="noopener">Cloudflare Cookie-Dokumentation</a>). Ein Verzicht auf Cloudflare-Bot-Mitigation würde das Sicherheits-Niveau der Verarbeitung personenbezogener Daten auf dieser Website schwächen. Daten: IP-Adresse, Request-Header, technischer Fingerprint der Anfrage. Kein persistenter, site-übergreifender Identifier. Speicherdauer: Cookie „__cf_bm“ läuft nach maximal 30 Minuten Inaktivität ab. Aggregierte Security-Logs bei Cloudflare werden gemäß deren Standard-Aufbewahrung kurzzeitig vorgehalten. Rechtsgrundlage: § 25 Abs. 2 Nr. 2 TTDSG (technisch erforderlich für den vom Nutzer gewünschten Dienst) sowie Art. 6 Abs. 1 lit. f DSGVO (überwiegendes berechtigtes Interesse an einer sicheren, verfügbaren Website). Drittlandübermittlung: Cloudflare ist ein US-Konzern; Daten können auf Servern in den USA verarbeitet werden. Garantien: EU-Standardvertragsklauseln und EU-US Data Privacy Framework (Angemessenheitsbeschluss vom 10. Juli 2023). Hinweis: Die Rechtssicherheit des Frameworks kann eingeschränkt sein; vor dem EuG ist ein Nichtigkeitsverfahren anhängig (Rechtssache T-553/23). Auftragsverarbeitung: Im Rahmen des Hosting-Vertrags geschlossen. Mehr Informationen:Cloudflare Datenschutzerklärung.
6.8 Google Search Console
Anbieter: Google Ireland Limited (siehe 6.1). Zweck: Google Search Console (GSC) ermöglicht es uns, das Verhalten unserer Website in den Google-Suchergebnissen zu überwachen — Indexierungs-Status, Such-Anfragen, die zur Seite führen, Klickraten und technische Crawl-Fehler. Cookies / Besucher-Tracking: GSC selbst setzt keine Cookies auf den Besuchern dieser Website und lädt kein clientseitiges Skript. Die Inhaberschaft der Domain ist über ein statisches HTML-Meta-Tag und / oder einen DNS-TXT-Eintrag verifiziert. Daten: Aggregierte Such-Analytics von Google (Suchanfragen, Impressionen, Klicks) — nicht direkt mit einzelnen Besuchern verknüpft. Uns werden die Daten nur in aggregierter Form bereitgestellt. Rechtsgrundlage: Art. 6 Abs. 1 lit. f DSGVO — berechtigtes Interesse an der Überwachung der Such-Sichtbarkeit. Drittlandübermittlung: Die aggregierten Reports werden auf Google-Infrastruktur (auch USA) erzeugt. Garantien wie unter 6.1. Mehr Informationen:Datenschutzerklärung Google.
6.9 Speicherdauer der Cookies
Session-Cookies werden beim Schließen des Browsers gelöscht. Persistente Cookies haben folgende maximale Laufzeiten: Google Analytics 4 — bis zu 14 Monate (Datenaufbewahrung in den GA4-Einstellungen auf 14 Monate verkürzt); Microsoft Clarity — bis zu 12 Monate; Google Ads („_gcl_au“) — bis zu 90 Tage; Meta-Pixel („_fbp“) — bis zu 90 Tage; LinkedIn („bcookie“) — bis zu 6 Monate; Cloudflare („__cf_bm“) — maximal 30 Minuten Inaktivität; Leadfeeder — in der Regel session-basiert, keine clientseitige Speicherung; CookieConsent (Cookiebot) — bis zu 12 Monate ab Erteilung der Einwilligung.
6.10 Widerruf der Einwilligung
So einfach wie die Erteilung — Sie können Ihre Einwilligung jederzeit mit Wirkung für die Zukunft widerrufen:
1. Cookie-Einstellungen öffnen: Klicken Sie auf den Link „Cookie-Einstellungen“ im Footer, um das Banner erneut zu öffnen und Ihre Auswahl anzupassen (steuert GA4, Clarity, Google Ads, LinkedIn, Meta-Pixel und Leadfeeder gleichermaßen). 2. Per E-Mail: Senden Sie eine kurze Nachricht an info@daniel-herrmann.io — wir blockieren die entsprechenden Tools für Sie. 3. Pro Tool: Für Google Analytics steht das offizielle Browser-Opt-out zur Verfügung; für Google Ads unter Werbe-Einstellungen anpassen; für LinkedIn unter LinkedIn-Opt-out; für den Meta-Pixel unter Meta Werbe-Einstellungen. Microsoft Clarity und Leadfeeder steuern Sie ausschließlich über unser Cookie-Banner.
Die Rechtmäßigkeit der bis zum Widerruf auf Grundlage der Einwilligung erfolgten Verarbeitung bleibt unberührt (Art. 7 Abs. 3 DSGVO).
7. Schriftarten
Diese Website lädt Schriftarten von Google Fonts. Hierbei werden Verbindungsdaten (insbesondere die IP-Adresse) an Google übermittelt. Anbieter: Google Ireland Limited (siehe Abschnitt 6.1). Rechtsgrundlage: Art. 6 Abs. 1 lit. f DSGVO — berechtigtes Interesse an einer einheitlichen typografischen Darstellung. Alternativ werden Schriftarten auch in einem lokalen Fallback vorgehalten, sodass das Schriftladen ausfallen kann, ohne das Layout zu brechen.
8. Technische & organisatorische Maßnahmen (TOM)
Zum Schutz Ihrer Daten setzen wir angemessene technische und organisatorische Maßnahmen gemäß Art. 32 DSGVO um — u. a.: TLS-Verschlüsselung (HTTPS) für die gesamte Seite, verschlüsselte Datenbankverbindungen, Server-Hosting in der EU (Kinsta / GCP Frankfurt), restriktiver Admin-Zugang über individuelle Accounts mit starken Passwörtern und 2FA, regelmäßige automatische Backups, rollenbasierte Zugriffskontrolle, ein dokumentiertes Verzeichnis von Verarbeitungstätigkeiten gemäß Art. 30 DSGVO und Datenminimierung auf Anwendungsebene. Wir überprüfen und aktualisieren diese Maßnahmen kontinuierlich.
9. Ihre Rechte als betroffene Person
Ihnen stehen nach der DSGVO folgende Rechte zu:
Recht auf Auskunft (Art. 15 DSGVO) — Auskunft über die zu Ihrer Person verarbeiteten Daten.
Recht auf Berichtigung (Art. 16 DSGVO) — Korrektur unrichtiger Daten.
Recht auf Löschung (Art. 17 DSGVO) — Löschung Ihrer Daten, soweit die gesetzlichen Voraussetzungen vorliegen.
Recht auf Einschränkung der Verarbeitung (Art. 18 DSGVO).
Recht auf Datenübertragbarkeit (Art. 20 DSGVO) — Erhalt Ihrer Daten in einem strukturierten, gängigen, maschinenlesbaren Format.
Recht auf Widerspruch (Art. 21 DSGVO) — gegen Verarbeitungen auf Basis berechtigter Interessen, insbesondere gegen Direktwerbung. Siehe hervorgehobener Hinweis unten.
Recht auf Widerruf erteilter Einwilligungen (Art. 7 Abs. 3 DSGVO) — jederzeit mit Wirkung für die Zukunft, siehe Abschnitt 6.10.
Beschwerderecht (Art. 77 DSGVO) — bei einer Aufsichtsbehörde, insbesondere im EU-Mitgliedstaat Ihres Aufenthalts, Arbeitsplatzes oder Ortes des mutmaßlichen Verstoßes. Für unseren Sitz zuständig: Unabhängiges Datenschutzzentrum Saarland, Fritz-Dobisch-Straße 12, 66111 Saarbrücken.
Anfragen zu diesen Rechten richten Sie bitte an: info@daniel-herrmann.io. Wir antworten innerhalb der gesetzlichen Frist (in der Regel ein Monat).
10. Aktualität dieser Datenschutzerklärung
Stand: 29. Juni 2026. Wir behalten uns vor, diese Erklärung anzupassen, damit sie stets den aktuellen rechtlichen Anforderungen entspricht. Die jeweils aktuelle Fassung ist über diese Website abrufbar.
Rückruf
Wann sollen wir Sie zurückrufen?
Sagen Sie uns, wann Sie Zeit haben und worum es geht — wir rufen innerhalb eines Werktags zurück.
Notiert. Wir melden uns.
Sie bekommen gleich eine Bestätigungs-E-Mail. Daniel ruft innerhalb eines Werktags zurück.
Per E-Mail erhalten
Wir schicken es Ihnen zu.
Name und E-Mail genügen — wir schicken Ihnen das Material direkt mit einer kurzen Notiz von Daniel.
Danke — bitte schauen Sie in Ihr Postfach.
Wir haben Ihnen das Material gerade per E-Mail geschickt. Sollte es nicht in den nächsten Minuten ankommen, schauen Sie bitte im Spam-Ordner nach.
Einmal im Monat · CSV aus der Praxis
Validierungs-Insights aus echten Pharma-Projekten.
Was Sie als nächstes lesen: was die GAMP 5 2nd Edition für Ihre CSV bedeutet, wie die FDA CSA Final Guidance Ihre Validierungsstrategie verändert, und der 8-Wochen-Sprint, mit dem wir 60+ Projekte abschließen. Jederzeit mit einem Klick abbestellbar.
Fast geschafft.
Bestätigen Sie Ihre E-Mail über den Link, den wir Ihnen gerade ins Postfach geschickt haben. Direkt nach der Bestätigung senden wir Ihnen die aktuelle Newsletter-Ausgabe.
Wir nutzen Ihre Daten ausschließlich für den Newsletter. Keine Weitergabe an Dritte. Datenschutzhinweis.