
CSA Methodology: Risk-Based Validation Explained in 5 Steps
Implement Computer Software Assurance concretely: the 5 steps of CSA methodology following FDA guidance. 60+ projects, 100% audit pass rate.
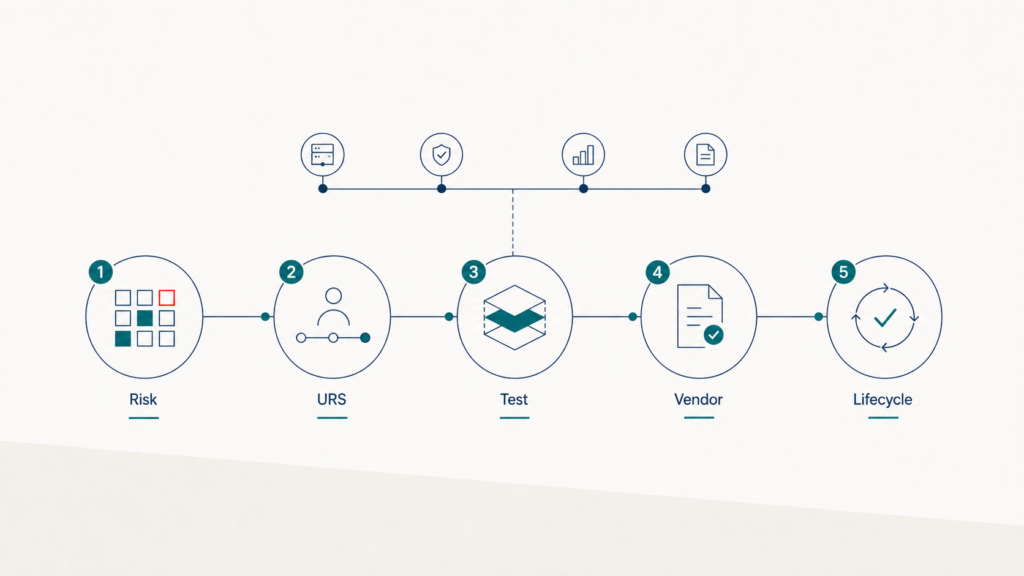
Computer Software Assurance (CSA) ist der von der FDA geprägte Ansatz für risikobasierte Software Assurance im regulierten Umfeld. Statt jede Funktion gleich tief zu testen, fokussieren Sie den Aufwand auf hochrisiko-relevante Features. Fünf Schritte führen durch die Implementierung: Risikobewertung, Use-Case-Zentrierung, gestaffelte Testtiefe, Vendor Evidence und kontinuierliche Anpassung. CSA ersetzt CSV nicht — sie schärft die Methodik im Sinne der FDA-Guidance zu Computer Software Assurance und der GAMP 5 Second Edition. In unseren Projekten reduziert sich der Validierungsaufwand dadurch typischerweise deutlich — bei gleichbleibender Audit-Belastbarkeit.
Ihr Team validiert ein neues System nach einem etablierten CSV-Framework. Alle Funktionen werden ähnlich tief getestet, die Dokumentation wächst, das Go-Live-Datum rückt nach hinten. Im Steering wird gefragt, warum es nicht schneller geht — obwohl jeder im Raum weiß, dass ein großer Teil der Tests auf Funktionen entfällt, die niemand kritisch nutzt.
Die FDA hat mit Computer Software Assurance (CSA) einen Ansatz geprägt, der genau hier ansetzt. CSA fokussiert Test-Aufwand auf das, was wirklich zählt: hohes Risiko. In der Praxis wird CSA jedoch oft missverstanden — als „weniger validieren” interpretiert, statt als „klüger validieren”. Genau dort entstehen die neuen Audit-Risiken.
Dieser Leitfaden zeigt die fünf Schritte einer sauberen CSA-Umsetzung — und wo die häufigsten Missverständnisse lauern.
Was CSA wirklich ist (und was nicht)
Computer Software Assurance ist ein risikobasierter Validierungs-Ansatz, den die FDA mit ihrer Guidance zu Computer Software Assurance fachlich geschärft hat. Der Kern: Critical Thinking First — Dokumentation folgt dem Risiko, nicht umgekehrt.
Moderne CSA-Ansätze ermöglichen es, Test-Aufwand und Dokumentation stärker auf risikorelevante Funktionen zu fokussieren. Statt jedes Feature gleich tief zu dokumentieren, jeden Workflow zu testen und jede Konfiguration zu belegen, lenkt CSA Aufmerksamkeit gezielt dorthin, wo Risiko entsteht. So entstehen schlankere Validierungs-Pakete — und Teams gewinnen Zeit für risikoorientiertes Denken.
CSA dreht die Logik um. Die Frage lautet nicht mehr „Was müssen wir alles testen?”, sondern „Wo kann ein Fehler echten Schaden verursachen — und wie reduzieren wir genau dieses Risiko?”.
Legacy-CSV-Umsetzung vs. moderner CSA-Ansatz
| Aspekt | Legacy-CSV-Umsetzung | Moderner CSA-Ansatz |
|---|---|---|
| Test-Strategie | Volltestung aller Funktionen | Risikobasiert: Fokus auf hohes Risiko |
| Dokumentations-Output | typischerweise 200–500 Seiten | typischerweise 60–120 Seiten (audit-belastbar) |
| Vendor-Hebel | eigene Tests dominant | Vendor Evidence strukturiert eingebunden |
| Tempo | erfahrungsgemäß 6–14 Monate | in vielen Projekten 8–12 Wochen |
| Mindset | Dokumentation first | Critical Thinking first |
| FDA-/Guidance-Bezug | anerkannt, aber oft dokumentationslastig umgesetzt | von der CSA-Guidance gestützt |
| Audit-Belastbarkeit | branchenüblich | gleich oder besser bei durchgängig risikobasierter Umsetzung |
Die 5 Schritte der CSA-Methodik
Schritt 1: Risikobewertung vor Test-Skript
Vor jedem Test steht die Frage: Was kann fehlgehen, wie wahrscheinlich ist es, wie schwerwiegend wäre es?
Die Bewertung erfolgt nach ICH Q9 — strukturiert, dokumentiert, nachvollziehbar. Jede Funktion erhält eine Risiko-Klassifikation: hoch, mittel, niedrig. Diese Klassifikation steuert ab sofort jeden weiteren Schritt.
Praktischer Output: Eine Risiko-Matrix pro System. Für ein typisches LIMS sind das 15–30 funktionale Bereiche, von denen meist 4–8 als High Risk eingestuft werden. Genau diese 4–8 Bereiche erhalten die volle Test-Tiefe.
Schritt 2: Use-Case-zentrierte Anforderungen
User Requirements Specifications (URS) werden in CSA nicht mehr als reine Feature-Liste geschrieben. Stattdessen beschreiben sie die kritischen User-Journeys: Welche Aufgaben muss der Nutzer im Alltag erfolgreich abschließen — und welche Fehler dürfen dabei nicht passieren?
Das schärft die URS in zwei Richtungen. Erstens: Sie wird kürzer und präziser. Zweitens: Sie liefert direkt die Testfälle, die später benötigt werden. In unseren Projekten reduziert eine gut geschriebene URS den späteren Validierungsaufwand spürbar.
Schritt 3: Gestaffelte Testtiefe nach Risiko
Statt jede Funktion mit dokumentierten Test-Skripten zu prüfen, wird die Testtiefe an das Risiko angepasst:
- High Risk: Ausführliche, dokumentierte Test-Skripte mit nachvollziehbarer Evidenz
- Medium Risk: Stichproben-Testung, ergänzt durch Vendor Evidence und dokumentierte Risikobegründung
- Low Risk: Reduzierte Testtiefe — abhängig von Intended Use, qualifiziertem Lieferantenstatus, GxP-Kritikalität und dokumentierter Risikobegründung. Eine Hersteller-Erklärung allein reicht nicht; die Bewertung muss begründet und nachvollziehbar sein.
Dieser Schritt erzeugt die größte Zeitersparnis. In unseren Projekten beobachten wir typischerweise 40–60 % weniger Test-Aufwand bei Standard-Systemen wie LIMS, MES oder ERP-Modulen.
Schritt 4: Vendor Evidence sauber strukturieren
Vendor Evidence ist mehr als „ISO-Zertifikat akzeptieren”. Die FDA-Guidance erlaubt es, Lieferanten-Verantwortung als Teil der Validierungs-Evidenz zu nutzen — aber nur unter klaren Voraussetzungen:
- Lieferantenqualifizierung: Der Lieferant ist nach GxP-Kriterien bewertet und freigegeben
- Intended Use definiert: Was wird vom System erwartet, in welchem regulatorischen Kontext?
- GxP-Kritikalität klassifiziert: Welche regulatorische Bedeutung hat die jeweilige Funktion?
- Change Control vereinbart: Wie wird mit Lieferanten-Updates umgegangen?
- Traceability gesichert: Vendor Evidence ist eindeutig zur eigenen Risikobewertung verknüpft
- Evidenz-Bewertung dokumentiert: Hersteller-Tests werden nicht ungeprüft übernommen, sondern bewertet
Mit diesem Setup ersetzen qualifizierte Lieferanten-Belege ganze Test-Pakete — bei nachvollziehbarer Audit-Belastbarkeit.
Schritt 5: Kontinuierliche Anpassung im Lifecycle
CSA ist kein einmaliger Setup-Akt vor dem Go-Live. Sie ist ein kontinuierlicher Prozess. Nach Go-Live werden Risiko-Bewertungen regelmäßig überprüft — typischerweise jährlich oder bei wesentlichen Änderungen.
Bei Patches, Updates oder neuen Integrations-Punkten wird das Risiko-Profil neu bewertet, nicht das gesamte System neu validiert. Das hält den laufenden Validierungs-Aufwand niedrig und sichert gleichzeitig die kontinuierliche Compliance.
Governance: Die unsichtbare Voraussetzung
CSA funktioniert nur, wenn IT, QA, Fachbereich und Lieferant dieselbe Risiko-Logik verwenden. Eine isolierte Methodik-Umstellung in der IT-Abteilung greift nicht — die Risiko-Bewertung muss als gemeinsamer Bewertungsstandard zwischen allen Stakeholdern verankert werden.
Praktisch bedeutet das: Ein gemeinsamer Risk-Assessment-Standard, klare Schnittstellen-Vereinbarungen, ein abgestimmtes Change-Management und regelmäßige Stakeholder-Reviews. Wo dieser Konsens fehlt, produziert CSA Reibungsverluste statt Effizienz.
Drei häufige Missverständnisse — und wie Sie sie vermeiden
Missverständnis 1: „CSA bedeutet weniger Tests.”
CSA bedeutet gezieltere Tests. Bei hochrisiko-relevanten Funktionen wird sogar tiefer getestet — weil die Aufmerksamkeit gezielt dort konzentriert wird. Wer CSA als Spar-Programm interpretiert, produziert genau die Audit-Findings, die der Ansatz eigentlich verhindern soll.
Missverständnis 2: „CSA ersetzt CSV.”
CSA ist eine Weiterentwicklung der CSV-Methodik, kein Ersatz. Beide Frameworks koexistieren. Hochkritische Sicherheits-Systeme (etwa Pharmakovigilanz oder klinische Datenbanken) bleiben in der Regel im klassischen CSV-Pfad. Standard-Systeme — LIMS, MES, ERP-Module — profitieren am stärksten von CSA.
Missverständnis 3: „CSA gilt nur für den FDA-Markt.”
Die FDA hat CSA initiiert. Auch europäische GxP-Erwartungen stützen risikobasiertes Vorgehen, etwa über Annex 11 und GAMP 5 Second Edition. CSA wird damit zunehmend zur tragenden Methodik in DACH-Pharma und Biotech — unabhängig davon, ob der primäre Markt USA oder EU ist.
Was Sie als IT-Leiter konkret tun können
Drei Hebel, in dieser Reihenfolge:
1. Quick Win (4–6 Wochen): Eine retrospektive Risiko-Bewertung Ihres aktiv validierten Systems. Identifiziert, welche Test-Pakete im nächsten Validierungs-Zyklus reduziert werden können — bei voller Audit-Belastbarkeit.
2. Mid-Term (3–6 Monate): URS-Modernisierung für das nächste Validierungs-Projekt. Use-Case-zentrierte Spezifikationen reduzieren den späteren Aufwand systematisch.
3. Long-Term (6–12 Monate): Team-Skills und Governance aufbauen. CSA verlangt risikobasiertes Denken auf jeder Ebene — vom Tester bis zum QA-Manager. Schulungen und Coaching während eines echten Projekts wirken deutlich besser als isolierte Trainings. Parallel: gemeinsamer Risk-Assessment-Standard für IT, QA, Fachbereich und Lieferanten.
Häufige Fragen
Eignet sich CSA für alle GxP-Systeme?
Nein. Für hochkritische Sicherheits-Systeme — etwa Pharmakovigilanz-Plattformen oder klinische Datenbanken — bleibt klassisches CSV der Standard. Für viele Standard-Systeme wie LIMS, MES, ERP-Module und Cloud-Plattformen bietet CSA einen praxistauglichen, regulatorisch anschlussfähigen Weg.
Wie steht CSA zur GAMP 5 Second Edition?
Sehr gut kompatibel. GAMP 5 Second Edition stützt risikobasierte Validierung methodisch und ist mit der CSA-Logik gut vereinbar. Beide Frameworks lassen sich ohne Konflikt parallel anwenden.
Was kostet die Umstellung auf CSA?
Der Aufwand hängt vom Reifegrad Ihrer aktuellen Validierungs-Praxis ab. Ein Strategiegespräch klärt in 30 Minuten, wo Ihr aktuelles Validierungs-Setup steht und welche CSA-Hebel realistisch sind.
Quellen
- FDA Guidance zu Computer Software Assurance (Final)
- ISPE GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems, Second Edition (Juli 2022)
- ICH Q9(R1) Quality Risk Management
- EU-GMP Annex 11
- 21 CFR Part 11: Electronic Records; Electronic Signatures
Autor
Daniel Herrmann Consulting — Boutique-Beratung für GxP-Compliance und Computer System Validation in Pharma, Biotech und MedTech. 15+ Jahre Hands-on-Expertise. 60+ validierte Systeme. 100 % Audit-Bestehensquote. 0 kritische Findings.
Computer Software Assurance (CSA) is the FDA-shaped approach to risk-based software assurance in regulated environments. Instead of testing every function with the same depth, you focus effort on high-risk-relevant features. Five steps guide the implementation: risk assessment, use-case centring, tiered test depth, vendor evidence, and continuous adaptation. CSA does not replace CSV — it sharpens the methodology in line with the FDA Guidance on Computer Software Assurance and GAMP 5 Second Edition. In our projects, this typically reduces validation effort significantly — at consistent audit reliability.
Lead
Your team validates a new system following an established CSV framework. All functions are tested at similar depth, documentation grows, the go-live date keeps slipping. In the steering committee, the question is raised why progress is slow — even though everyone in the room knows that a large share of the tests covers functions no one uses critically.
The FDA has shaped an approach with Computer Software Assurance (CSA) that addresses exactly this. CSA focuses test effort on what really matters: high risk. In practice, however, CSA is often misunderstood — interpreted as "validate less" instead of "validate smarter". That is precisely where new audit risks arise.
This guide shows the five steps of a clean CSA implementation — and where the most common misconceptions lie.
What CSA Really Is (and What It Is Not)
Computer Software Assurance is a risk-based validation approach that the FDA has sharpened technically with its Guidance on Computer Software Assurance. The core: Critical Thinking First — documentation follows risk, not the other way around.
Modern CSA approaches allow you to focus test effort and documentation more strongly on risk-relevant functions. Instead of documenting every feature to the same depth, testing every workflow and substantiating every configuration, CSA directs attention specifically to where risk arises. The result: leaner validation packages — and teams gain time for risk-oriented thinking.
CSA flips the logic. The question is no longer "What do we need to test?", but "Where can a defect cause real damage — and how do we mitigate exactly that risk?"
Legacy CSV Implementation vs. Modern CSA Approach
| Aspect | Legacy CSV Implementation | Modern CSA Approach |
|---|---|---|
| Test strategy | Full testing of all functions | Risk-based: focus on high risk |
| Documentation output | Typically 200–500 pages | Typically 60–120 pages (audit-reliable) |
| Vendor leverage | Own tests dominant | Vendor evidence structurally integrated |
| Pace | Empirically 6–14 months | In many projects 8–12 weeks |
| Mindset | Documentation first | Critical thinking first |
| FDA / guidance link | Recognised, but often implemented documentation-heavy | Supported by the CSA guidance |
| Audit reliability | Industry standard | Equal or better with consistently risk-based implementation |
The 5 Steps of CSA Methodology
Step 1: Risk Assessment Before Test Script
Before any test comes the question: What can go wrong, how likely is it, how severe would it be?
The assessment follows ICH Q9 — structured, documented, traceable. Each function receives a risk classification: high, medium, low. This classification governs every subsequent step.
Practical output: A risk matrix per system. For a typical LIMS, that means 15–30 functional areas, of which 4–8 are usually classified as high risk. Exactly these 4–8 areas receive full test depth.
Step 2: Use-Case-Centred Requirements
User Requirements Specifications (URS) in CSA are no longer written as pure feature lists. Instead, they describe critical user journeys: Which tasks must the user complete successfully in daily work — and which errors must not occur?
This sharpens the URS in two ways. First: it becomes shorter and more precise. Second: it directly yields the test cases needed later. In our projects, a well-written URS noticeably reduces subsequent validation effort.
Step 3: Tiered Test Depth by Risk
Instead of checking each function with documented test scripts, test depth is matched to risk:
- High risk: Comprehensive, documented test scripts with traceable evidence
- Medium risk: Sampling-based testing, supplemented by vendor evidence and documented risk justification
- Low risk: Reduced test depth — depending on intended use, qualified vendor status, GxP criticality and documented risk justification. A manufacturer's declaration alone is not sufficient; the assessment must be justified and traceable.
This step delivers the greatest time savings. In our projects, we typically observe 40–60 % less test effort on standard systems such as LIMS, MES or ERP modules.
Step 4: Structuring Vendor Evidence Cleanly
Vendor evidence is more than "accept an ISO certificate". The FDA guidance allows you to use vendor responsibility as part of validation evidence — but only under clear conditions:
- Vendor qualification: The vendor is assessed and approved against GxP criteria
- Intended use defined: What is expected from the system, in which regulatory context?
- GxP criticality classified: What regulatory significance does each function have?
- Change control agreed: How are vendor updates handled?
- Traceability ensured: Vendor evidence is unambiguously linked to your own risk assessment
- Evidence assessment documented: Vendor tests are not adopted unexamined but evaluated
With this setup, qualified vendor evidence replaces entire test packages — with traceable audit reliability.
Step 5: Continuous Adaptation across the Lifecycle
CSA is not a one-time setup before go-live. It is a continuous process. After go-live, risk assessments are reviewed regularly — typically annually or on significant changes.
For patches, updates or new integration points, the risk profile is reassessed, not the entire system revalidated. This keeps ongoing validation effort low and ensures continuous compliance at the same time.
Governance: The Invisible Prerequisite
CSA only works if IT, QA, business and vendor use the same risk logic. An isolated methodology shift in IT alone does not take hold — the risk assessment must be anchored as a shared evaluation standard across all stakeholders.
In practice, that means a shared risk-assessment standard, clear interface agreements, coordinated change management and regular stakeholder reviews. Where this consensus is missing, CSA generates friction instead of efficiency.
Three Common Misconceptions — and How to Avoid Them
Misconception 1: "CSA means fewer tests."
CSA means more targeted tests. On high-risk-relevant functions, it actually tests deeper — because attention is concentrated there deliberately. Anyone who interprets CSA as a savings programme produces exactly the audit findings the approach is designed to prevent.
Misconception 2: "CSA replaces CSV."
CSA is an evolution of CSV methodology, not a replacement. Both frameworks coexist. Highly critical safety systems (e.g. pharmacovigilance or clinical databases) usually remain on the classic CSV path. Standard systems — LIMS, MES, ERP modules — benefit the most from CSA.
Misconception 3: "CSA only applies to the FDA market."
The FDA initiated CSA. European GxP expectations also support risk-based approaches, for example via Annex 11 and GAMP 5 Second Edition. CSA is therefore increasingly becoming a load-bearing methodology in DACH pharma and biotech — regardless of whether the primary market is the US or the EU.
What You Can Do Concretely as IT Lead
Three levers, in this order:
1. Quick win (4–6 weeks): A retrospective risk assessment of your actively validated system. Identifies which test packages can be reduced in the next validation cycle — at full audit reliability.
2. Mid-term (3–6 months): URS modernisation for the next validation project. Use-case-centred specifications systematically reduce later effort.
3. Long-term (6–12 months): Build team skills and governance. CSA demands risk-based thinking at every level — from tester to QA manager. Training and coaching during a real project work significantly better than isolated trainings. In parallel: a shared risk-assessment standard for IT, QA, business and vendors.
Frequently Asked Questions
Is CSA suitable for all GxP systems?
No. For highly critical safety systems — such as pharmacovigilance platforms or clinical databases — classic CSV remains the standard. For many standard systems such as LIMS, MES, ERP modules and cloud platforms, CSA offers a practicable, regulatorily compatible path.
How does CSA relate to GAMP 5 Second Edition?
Very compatible. GAMP 5 Second Edition supports risk-based validation methodologically and aligns well with CSA logic. Both frameworks can be applied in parallel without conflict.
What does the transition to CSA cost?
Effort depends on the maturity of your current validation practice. A strategy call clarifies in 30 minutes where your current validation setup stands and which CSA levers are realistic.
Sources
- FDA Guidance on Computer Software Assurance (Final)
- ISPE GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems, Second Edition (July 2022)
- ICH Q9(R1) Quality Risk Management
- EU-GMP Annex 11
- 21 CFR Part 11: Electronic Records; Electronic Signatures
Author
Daniel Herrmann Consulting — boutique consultancy for GxP compliance and Computer System Validation in pharma, biotech and MedTech. 15+ years of hands-on expertise. 60+ validated systems. 100 % audit pass rate. 0 critical findings.